This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison.
OverviewTay-Sachs disease (TSD) is an autosomal recessive disorder caused by a mutation that leaves the body unable to produce an enzyme known as hexosaminidase-A (Hex-A) [1]. Hex-A is necessary for fat metabolism in nerve cells. By the absence of this enzyme, central nervous system degeneration ensues due to the accumulation of lipid called GM2 ganglioside in the nerve cells of the brain [2].
|
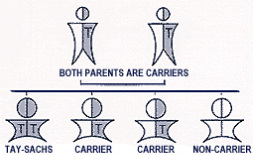
Figure 1: Tay-Sachs occurs when both parents carry a Tay-Sachs gene and each parent transmits the defective gene to their child. (Figure from National Tay-Sachs & Allied Diseases Association of Delaware Valley)
|
Symptoms
There are basically three forms of Tay-Sachs, which are classic infantile, juvenile and late onset Tay-Sachs. They are determined based on the age of the affected individual when symptoms first appear.

Table 1: Types of Tay-Sachs by the age when the first symptoms first appear on affected individual.
(Information adapted from National Tay-Sachs & Allied Diseases)
|
Infants with classic infantile TSD appear normal until about the age of 3 to 6 months. They start to lose motor skills such as crawling, turning over, and sitting. By the age of two, the affected children experience seizures. As the disease progresses, other symptoms include vision and hearing loss, intellectual disability, and paralysis. They also develop an exaggerated startle response to noise[2]. Besides, an eye abnormality called a cherry-spot is another characteristic of this disorder [3]. Children with this severe infantile form of TSD usually live only into early childhood.
Juvenile forms of TSD are rare. Children appear normal until middle childhood. People often dismissed the clumsiness which is normally the first sign of this disorder. Over time, the affected children begin to show the symptoms as infants with classic infantile Tay-Sachs. Most of those with juvenile forms of Tay-Sachs die at the age around 15 [4]. Late-onset TSD is a much rarer form of Tay-Sachs, which occurs in person who has a genetic mutation that is similar. Those signs and symptoms shown by other types of Tay-Sachs vary widely among people with late-onset forms of TSD [5]. |
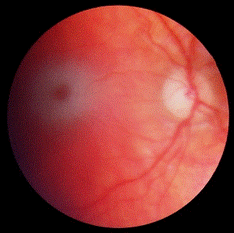
Figure 2: A “cherry-red” spot appears in the back of the eyes, a symptom of Tay-Sachs. (Figure from http://flipper.diff.org/app/items/info/2950)
|
|

Figure 3: HEXA gene (yellow arrow) is on chromosome 15. (Figure from Genetics Home Reference)
|
Prevalence of Tay-Sachs Disease
- The carrier rate of the HEXA mutation in general population is estimated one in 300. (Natural Standard: The Authority on Integrative Medicine)
- However, TSD is not common in the general population. In terms of geographical distribution, the incidence of Tay-Sachs is more common among people of Eastern European, Central European and Askhenazi Jewish heritage [7].
- Before the introduction of genetic screening, Non-Jewish French Canadian and the Cajun community of Louisiana had the same carrier frequency of the TSD gene as Ashkenazi Jews populations (about 1 in 27) [7].
- The carrier rate among the Irish Americans is about one in 50.
- However, TSD is not common in the general population. In terms of geographical distribution, the incidence of Tay-Sachs is more common among people of Eastern European, Central European and Askhenazi Jewish heritage [7].
- Before the introduction of genetic screening, Non-Jewish French Canadian and the Cajun community of Louisiana had the same carrier frequency of the TSD gene as Ashkenazi Jews populations (about 1 in 27) [7].
- The carrier rate among the Irish Americans is about one in 50.
Current treatment
Identification of mutations in the HEXA gene and the recognition of enzymatic activity of Hex A have been practiced in prenatal diagnosis and clinical diagnosis of people with TSD. Nevertheless, currently, there is no effective cure that will prevent TSD. Treatments are mainly focus on controlling the symptoms and care issues [6].

Table 2: Treatments for Tay-Sachs. (Information derived from Mayo Clinic)
Apart from these treatments, researchers have attempted to investigate novel therapies for TSD. One of the experimental treatments is the utility of enzyme replacement therapy (ERT), which delivers Hex-A enzyme to the patient’s brain [8].
Another avenue of research is gene therapy, which involves the transfer of functional copies of HEXAgene into TSD patients. However, the effectiveness of this therapy has not been verified. In this case, the discovery of TSD in Jacob sheep is helpful to further investigating the utility of gene therapy for treatment of TSD. This is because the biochemical mechanisms of the Jacob sheep model of TSD are quite identical to the human classic infantile form of TSD. Hence, it can be a useful tool for trials of gene therapy [9].
Besides, stem cell therapy can be a potential treatment for TSD but it is still unproven by American medical standards (Tampa Bay Times). The video (left) is the news about a little girl with TSD, who is only 16 months old, underwent two rounds of Stem Cell Treatment.
Another avenue of research is gene therapy, which involves the transfer of functional copies of HEXAgene into TSD patients. However, the effectiveness of this therapy has not been verified. In this case, the discovery of TSD in Jacob sheep is helpful to further investigating the utility of gene therapy for treatment of TSD. This is because the biochemical mechanisms of the Jacob sheep model of TSD are quite identical to the human classic infantile form of TSD. Hence, it can be a useful tool for trials of gene therapy [9].
Besides, stem cell therapy can be a potential treatment for TSD but it is still unproven by American medical standards (Tampa Bay Times). The video (left) is the news about a little girl with TSD, who is only 16 months old, underwent two rounds of Stem Cell Treatment.
References
1. Myerowitz, R. (1997). Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene. Human Mutation, 9(3), 195-208. Retrieved February 1, 2012, from http://onlinelibrary.wiley.com.ezproxy.library.wisc.edu/doi/10.1002/(SICI)1098-1004(1997)9:3%3C195::AID-HUMU1%3E3.0.CO;2-7/pdf
2. National Human Genome Research Institute. (2011). Learning about Tay-Sachs Disease. Retrieved February 6, 2012, from http://www.genome.gov/page.cfm?pageID=10001220#what
3. Leavitt, J. , & Kotagal, S. (2007). The "cherry red" spot. Pediatric Neurology, 37(1), 74-75. doi: 10.1016/j.pediatrneurol.2007.04.011
4. National Tay-Sachs & Allied Diseases. The Disease: Tay-Sachs Disease. Retrieved February 6,2012, from http://www.ntsad.org/index.php/tay-sachs
5. Aetna InteliHealth. (2011). Tay-Sachs genetic testing basics. Retrieved January 28, 2012, from http://intelihealth.com/IH/ihtIH/WSIHW000/32193/35422.html
6. B.J. Zeng, P.A. Torres, T.C. Viner, Z.H. Wang, S.S. Raghavan, J. Alroy, G.M. Pastores & E.H. Kolodny. (2008). Spontaneous appearance of Tay–Sachs disease in an animal model. Molecular Genetics and Metabolism, 95(1-2), 59-65. doi: 10.1016/j.ymgme.2008.06.010
7. World Health Organization. (2012). Electronic reference. Retrieved February 7, 2012, from http://www.who.int/genomics/public/geneticdiseases/en/index2.html
8. Matsuoka, K., Tamura, T., Tsuji, D., Dohzono Y., Kitakaze K., et al. (2011).Therapeutic potential of intracerebroventricular replacement of
modified human β-Hexosaminidase B for GM2 gangliosidosis. Molecular Therapy, 19(6), 1017–1024. doi: 10.1038/mt.2011.27
9. Torres, P. , Zeng, B. , Porter, B. , Alroy, J. , Horak, F. , et al. (2010). Tay-sachs disease in Jacob sheep. Molecular Genetics & Metabolism, 101(4),
357-363. doi: 10.1016/j.ymgme.2010.08.006
2. National Human Genome Research Institute. (2011). Learning about Tay-Sachs Disease. Retrieved February 6, 2012, from http://www.genome.gov/page.cfm?pageID=10001220#what
3. Leavitt, J. , & Kotagal, S. (2007). The "cherry red" spot. Pediatric Neurology, 37(1), 74-75. doi: 10.1016/j.pediatrneurol.2007.04.011
4. National Tay-Sachs & Allied Diseases. The Disease: Tay-Sachs Disease. Retrieved February 6,2012, from http://www.ntsad.org/index.php/tay-sachs
5. Aetna InteliHealth. (2011). Tay-Sachs genetic testing basics. Retrieved January 28, 2012, from http://intelihealth.com/IH/ihtIH/WSIHW000/32193/35422.html
6. B.J. Zeng, P.A. Torres, T.C. Viner, Z.H. Wang, S.S. Raghavan, J. Alroy, G.M. Pastores & E.H. Kolodny. (2008). Spontaneous appearance of Tay–Sachs disease in an animal model. Molecular Genetics and Metabolism, 95(1-2), 59-65. doi: 10.1016/j.ymgme.2008.06.010
7. World Health Organization. (2012). Electronic reference. Retrieved February 7, 2012, from http://www.who.int/genomics/public/geneticdiseases/en/index2.html
8. Matsuoka, K., Tamura, T., Tsuji, D., Dohzono Y., Kitakaze K., et al. (2011).Therapeutic potential of intracerebroventricular replacement of
modified human β-Hexosaminidase B for GM2 gangliosidosis. Molecular Therapy, 19(6), 1017–1024. doi: 10.1038/mt.2011.27
9. Torres, P. , Zeng, B. , Porter, B. , Alroy, J. , Horak, F. , et al. (2010). Tay-sachs disease in Jacob sheep. Molecular Genetics & Metabolism, 101(4),
357-363. doi: 10.1016/j.ymgme.2010.08.006
|
|
